
‘Field action’ announced for LIFEPAK® 15 Monitor/ Defibrillator
Three companies have issued recalls this week, two for medications and one for a medical device.
Lamotrigine Tablets
Taro Pharmaceuticals U.S.A., Inc. on Jan. 10 issued a voluntarily nation-wide recall of one lot of Lamotrigine 100 mg tablets to the consumer level.
This single lot of Lamotrigine 100 mg Tablets Lot #331771 (expiration date June 2021) was found to have been cross-contaminated with a small amount of another drug substance (Enalapril Maleate) used to manufacture another product at the same facility.
Lamotrigine 100 mg Tablets are indicated for Epilepsy and Bipolar disorder. This product is packaged in white plastic bottles with screw cap closure, and each bottle contains 100 tablets. Each bottle is labeled to indicate the name of the product, Lamotrigine Tablets USP, 100 mg, the NDC #51672-4131-1 (see image of container label below), the lot number 331771 and expiration date of June 2021.

Use of Lamotrigine 100 mg Tablets could potentially result in exposure to a small amount of Enalapril Maleate, if present in the product in question
Enalapril Maleate is a drug substance indicated for hypertension and congestive heart failure. There is potential with chronic exposure to Enalapril Maleate to impact users particularly if they are small children or pregnant women. Enalapril Maleate is also associated with risk of birth defects in a developing fetus. Therefore, there is risk associated with the continued, long-term use of that lot of Lamotrigine 100 mg tablets.
Lamotrigine 100 mg Tablets, Lot # 331771 were distributed to wholesale distributors in the US market between Aug. 23 and Aug. 30, 2019.
Taro is notifying its distributors and customers by Phone, E-mail, and Letters via US Mail and is arranging for return of any containers or quantities of Lamotrigine 100 mg Tablets, Lot # 331771 (exp. June 2021). The company recommends consumers who have any quantities of Lamotrigine 100 mg Tablets, Lot # 331771 to stop using this product and return it to the pharmacy that dispensed it.
Consumers with questions regarding this recall can contact Taro by calling 1-866-923-4914 or by e-mail at [email protected], Monday through Friday between 7 a.m. and 7 p.m. US Central Time. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Nizatidine Capsules
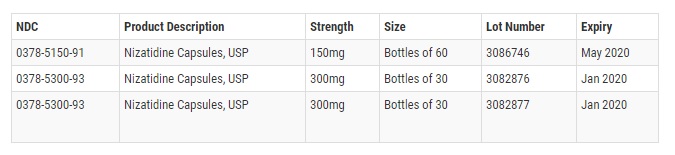
Mylan Pharmaceuticals business Jan. 8 announced a voluntary nationwide recall, to the consumer level, of three lots of Nizatidine Capsules, USP (including the 150 mg and 300 mg strengths), due to detected trace amounts of an impurity N-nitrosodimethylamine (NDMA).
NDMA is a known environmental contaminant and found in water and foods, including meats, dairy products and vegetables. NDMA has been classified as a probable human carcinogen (a substance that could cause cancer) according to the International Agency for Research on Cancer (IARC).
These batches were distributed nationwide to wholesalers, mail order pharmacies, retail pharmacies, and a distributor between June 2017 and August 2018. The recalled products include:
Nizatidine is indicated for the short-term treatment (up to 8 weeks) of active duodenal ulcers and active benign gastric ulcers, as maintenance therapy for duodenal ulcer patients for up to one year, and for up to 12 weeks for the treatment of endoscopically diagnosed esophagitis and associated heartburn due to gastroesophageal reflux disease (GERD).
Mylan is notifying its distributors and customers by letter and is arranging for return of all recalled products. Wholesalers, retailers and consumers that are in possession of recalled product should contact Stericycle at 888-628-0727 for the return of the recalled product. Normal business hours are Monday through Friday 8 a.m. to 5 p.m. EST.
Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to using these drug products.


LIFEPAK 15 monitor/defibrillators
Stryker announced voluntary field action on specific units of the LIFEPAK 15 monitor/defibrillators.
The company is notifying a population of LIFEPAK 15 customers of an issue that may cause their devices to fail to deliver a defibrillation shock after the “Shock” button on the keypad is pressed. This is a result of oxidation that may have formed over time within the “Shock” button.
The company is contacting customers with impacted devices to schedule the correction of their device(s), which will include replacement of the affected keypad. Stryker anticipates that all devices subject to this field action will be serviced by June 2021.
Most complaints associated with this issue were detected prior to patient use. Routine testing of the device can detect this fault condition. If a customer experiences this issue, they should contact Stryker as soon as possible at 1-800-787-9537 and select option 2.
The company is instructing customers to continue to use their LIFEPAK 15 monitor/defibrillator according to the Operating Instructions until the correction can be completed. Customers should continue to perform the daily check as described in the Operator’s Checklist, specifically, the QUIK- COMBO therapy cable check as described in the General Maintenance and Testing Section (pages 10-4 and the LIFEPAK 15 monitor/defibrillator Operator’s Checklist, number 7).
Information about this notice is available at: http://www.strykeremergencycare.com/productnotices. Impacted customers will be notified by letter and will be requested to verify their device status.
Customers with questions regarding this notification, please contact Stryker by calling 1-800-787-9537, option 2, 8:00 A.M. to 7:00 P.M. (EST), Monday – Friday, or by email to [email protected] or 1-800- 329-7879.




